

ADC 临床剂量优化的一般考虑
发布时间:
2024-05-24 00:00
来源:

近年来,剂量优化已成为 FDA 审评肿瘤新药时重点关注的内容。
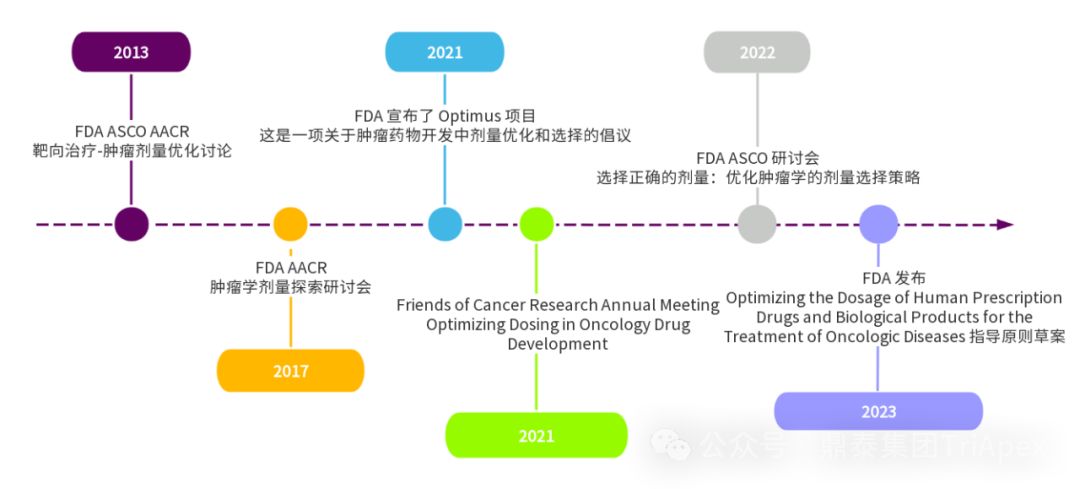
肿瘤药物临床剂量优化和 Project Optimus 项目产生历程
Project Optimus 是 FDA 肿瘤卓越中心(Oncology Centre of Excellence, OCE)发起的一项旨在改革肿瘤药物开发中剂量优化和剂量选择模式的项目。Project Optimus 团队由医学肿瘤学家、临床药理学家、药理学家/毒理学家、统计学家等多学科专家组成,旨在与工业界、学术界、专业协会、国际监管机构和患者合作,共同推进这一新的剂量优化范式。其主要目标可归纳如下:
-
教育和交流:通过指南、研讨会和其他公开会议,传达 FDA 对于剂量探索和剂量优化的期望。
-
早期互动:鼓励申请人在远早于拟用于注册的临床试验之前的早期开发阶段,与 FDA 肿瘤审评部门讨论剂量探索和剂量优化的策略。
-
制定新策略:利用非临床和临床数据制定剂量选择的新策略,该策略的重点是尽可能早期、高效地进行剂量优化,以便将有前景的新疗法可尽快惠及患者。
总的来说,Project Optimus 致力于改变传统的基于 MTD 的剂量选择模式,推动肿瘤药物开发中采用最大化疗效、安全性和耐受性的剂量优化策略。
目前,全球已有 15 款 ADC 药物上市,其中 7 款已经进入国内市场。在临床实践中,ADC 的应用仍受到在靶毒性、脱靶毒性和其他难以预知的不良反应的限制。由于毒性过大和不利的风险-获益状况,导致许多 ADC 药物在临床开发过程中失败。即使对于部分已获批的 ADC,很多患者由于无法耐受相关毒性,需要减少剂量、延迟或停止治疗。
在 ADC 药物的临床试验中,确定 FIH 起始剂量和在后续临床试验中进行剂量优化是至关重要的。鼎泰团队在对 FDA 发布的抗肿瘤药物剂量优化指南《Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases Guidance for Industry》和 ADC 药物临床药理学研究指南《Clinical Pharmacology Considerations for Antibody-Drug Conjugates Guidance for Industry》进行学习和理解的基础上,结合相关文献和案例探讨了 ADC 药物临床试验中进行起始剂量选择和剂量优化的一般考虑,以期能加深对本期主题的理解,为 ADC 药物临床研究提供参考。
★ 文章导览 ★
|
01 |
相关指导原则 |
|
02 |
ADC 药物的主要毒性和毒性来源 |
|
03 |
ADC 药物临床剂量优化建议 |
相关指导原则
1. 抗肿瘤药物剂量优化指南
2023年1月,FDA 发布了《Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases Guidance for Industry》[1],旨在帮助申请人在临床开发期间和提交新适应症及其用法用量申请之前,确定最佳剂量。
1.1 传统 MTD 范式(paradigm)的局限性
抗肿瘤药物的剂量探索试验(包括剂量递增和剂量扩展)常被用于 MTD 的确定。这种模式主要适用于细胞毒性化疗药物,基于观察到的陡峭的剂量(steep dose-response)反应关系、有限的靶标特异性,患者和医生有较高的接受毒性以治疗严重、危及生命的疾病的意愿。MTD 是通过短时间内在少数患者中逐步增加剂量直至观察到预先定义的严重或危及生命的 DLT 而确定的。在随后的临床试验中,通常使用 MTD 或接近 MTD 的剂量,而不进行进一步的剂量优化。
1.2 现代靶向治疗药物的特点
大多数现代肿瘤靶向治疗药物(如激酶抑制剂和单抗)设计为与肿瘤特异性信号通路的相互作用。与细胞毒性化疗相比,靶向治疗表现出不同的剂量-反应关系,低于 MTD 的剂量可能具有与 MTD 相似的疗效,但毒性更小。此外,在某些情况下可能永远无法达到 MTD。患者可能接受更长时间的靶向治疗,从而引起较低却持续存在的毒性。值得注意的是,在关键性注册临床试验中,通常采用 MTD 或剂量递增试验中的最高剂量。这种模式可能导致患者对 RP2D 的耐受性较差,对生活质量产生不利影响,并影响患者持续用药以获得最大的临床获益。此外,在一种治疗中出现 AE 的患者可能很难耐受后续治疗,尤其是在存在毒性叠加的情况下。
1.3 剂量优化的建议
传统的 MTD 范式通常无法对轻度症状毒性(1-2 级)、剂量调整、药物活性、剂量和暴露-反应关系以及特定人群(按年龄、器官损伤、合并用药或并发症进行定义)数据等进行评估。剂量探索试验可通过考察不同剂量下的临床数据、剂量-暴露反应关系选择值得进一步研究的剂量,是剂量优化的有效做法。以下对该指南中给出的剂量优化建议进行了总结,主要包括:
收集和解读临床药代动力学、药效学和药物基因组学数据
-
建议基于对相关非临床和临床数据的评估确定临床试验剂量
-
强调了收集和解读临床 PK、药效学和药物基因组学数据的重要性
-
推荐在剂量探索试验中包含 PK 采样和分析计划
-
强调在关键性临床试验中考虑特定人群的剂量差异
-
建议将相关协变量纳入 E-R 分析,以评估对不同亚组安全性和有效性可能的影响


比较多种剂量的试验设计
-
建议在临床试验中对多个剂量下的活性、安全性和耐受性进行比较以减少在 NDA 申请时最佳剂量选择的不确定性
-
推荐采用随机、平行剂量试验对不同剂量下的响应情况进行比较
-
多剂量对比试验通常在注册性临床试验之前完成,或在注册性试验中通过增加一个额外的队列开展

安全性和耐受性角度的考虑
-
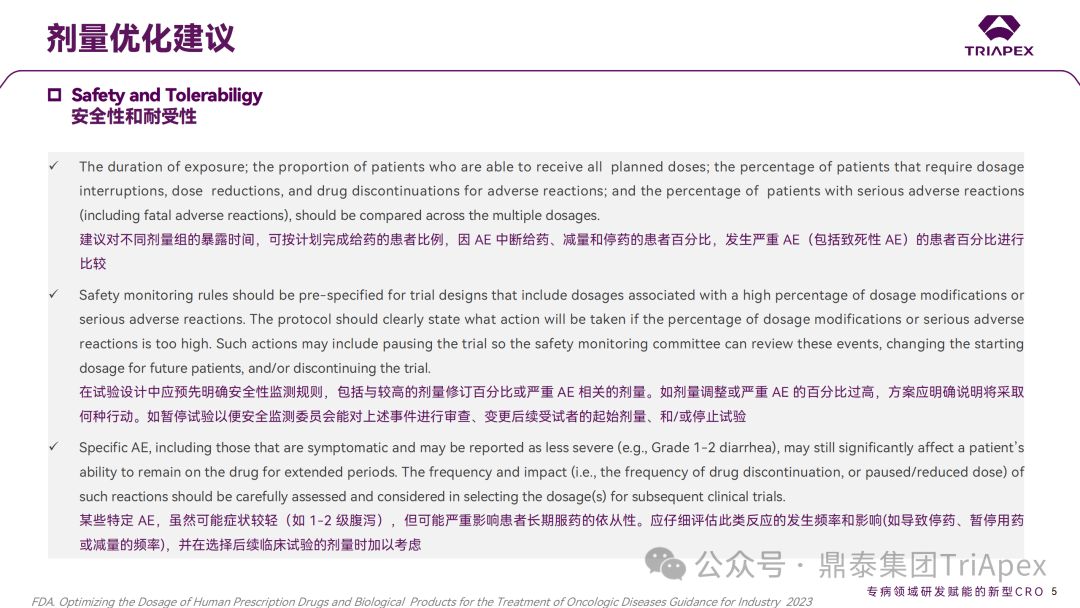
建议对不同剂量组的暴露时间,可按计划完成给药的患者比例,因 AE 中断给药、减量和停药的患者百分比,发生严重 AE(包括致死性 AE)的患者百分比进行比较
在试验设计中应预先明确安全性监测规则,包括与较高的剂量修订百分比或严重 AE 相关的剂量。如剂量调整或严重 AE 的百分比过高,方案应明确说明将采取何种行动,如暂停试验以便安全监测委员会能对上述事件进行审查、变更后续受试者的起始剂量、和/或停止试验
某些特定 AE,虽然可能症状较轻(如 1-2 级腹泻),但可能严重影响患者的长期用药。应仔细评估此类反应的发生频率和影响(如导致停药、暂停用药或减量的频率),并在选择后续临床试验的剂量时加以考虑
-
某些抗肿瘤药物可能与早发、严重或危及生命的毒性有关,这些毒性在后续给药后可能减轻或不再发生。这种情况下,可考虑使用替代给药策略的可行性,如逐步提高剂量(即滴定)以提高耐受性
-
患者报告结局(PRO)可以系统、定量地评估预期的症状性 AE 及其对功能的影响。在早期剂量探索试验中,应考虑纳入 PRO 以加强对耐受性的评估。关于 PRO 工具的选择和评估频率,建议参考相关指导原则
-
在选择最佳剂量时,与患者和其他关键利益相关方(如特定疾病领域的患者权益倡导组织)进行互动,有利于为最佳剂量的选择提供安全性和耐受性角度的参考

药物制剂
-
应提供不同的制剂规格,以便在临床试验中对多个剂量进行评估生产多种剂量规格的样品存在困难时,不足以成为不比较多个剂量的理由
-
对于口服给药,在选择最终剂型和规格时,应考虑单次剂量所需片剂或胶囊的大小和数量是否合适
-
对于肠外使用,在选择最终剂型和规格时,应考虑最终浓度和体积是否合适

新适应症和用法用量
-
根据肿瘤生物学、患者群体、治疗环境和合并用药(联合治疗)等因素的潜在差异,不同的疾病环境或肿瘤疾病可能需要不同的剂量。应考虑适用的非临床和临床数据,以支持在注册性临床试验中所选择的给药剂量和适用性,以支持后续的适应症和用法
在启动注册性临床试验之前,应提供所选择的剂量的充分理由,以支持后续的适应症和用法,特别是对于在前期剂量探索试验中没有充分探索过的瘤种或新的联合方案。如果不能提供剂量选择的充分理由,则应进行额外的剂量探索试验

2. ADC临床药理学指南
2022年2月,FDA 发布了《Clinical Pharmacology Considerations for Antibody-Drug Conjugates Guidance for Industry》[2],阐述了 FDA 目前对 ADC 药物开发过程中临床药理学相关的考虑和建议,包括生物分析方法、给药策略、剂量-暴露关系分析、内在因素、QTc 评估、免疫原性和药物-药物相互作用(DDI)。
以下重点对给药策略和剂量-暴露关系分析相关的内容进行梳理,以便总结与 ADC 药物临床剂量优化相关的思路,主要包括:
2.1 临床开发中的剂量选择
FDA 建议在早期药物开发中进行广泛的剂量范围探索试验,包括多种剂量水平和/或给药方案(如单次或多次给药),以充分描述 ADC 及其活性成分与安全性和活性之间的关系。建议根据早期临床研究结果进行 E-R 分析,以指导后期开发中选择合适的剂量优化策略。在适当的情况下,还应考虑固定剂量和基于体重的剂量。
2.2 针对内在和外在因素的剂量策略考虑
由于 ADC 的不同组分可能独立影响安全性和/或疗效,因此基于内在(如肾或肝功能不全)和外在因素(如药物相互作用)确定推荐剂量具有一定的挑战性。
调整特定人群的 ADC 剂量以实现与总体人群相似的某一组分(通常是 payload)的暴露,可能会导致 ADC 的暴露改变,进而影响疗效。应在 ADC开发过程中评估这些因素对 PK、安全性和有效性的影响,并在说明书中告知相关信息,以便为特定患者人群提供风险缓解策略。
2.3 临床药理学考虑
所有生物分析方法均应按照 FDA 指南进行验证和报告。从 FIH 开始,应采用经验证的分析方法检测 ADC 及其组分的浓度;后期临床试验中,建议检测 ADC 及其组分和可定量的药理活性代谢物,用于 E-R 分析。
如果未检测到脱偶联的 payload,可能不需要进行检测;如果 ADC 仅作为载体,且总抗体浓度与 ADC 高度相关,可能无需检测总抗体。
2.4 特定研究的考虑
在器官功能障碍人群的临床试验中,建议检测 ADC、脱偶联的 payload 和药理活性代谢物的水平。在 QTc 评估中,通常仅需检测脱偶联的 payload 和药理活性代谢物。在 DDI 研究中,如果脱偶联 payload 可检测,仅检测 paylaod 可能是足够的;如果抗体也可能参与 DDI,可能还需要检测 ADC 或总抗体。
2.5 E-R 分析
应针对 ADC 及其组分进行 E-R 分析,以支持剂量选择和调整。如果 ADC 的某个组分的系统暴露低,或者抗体无药理活性,或总抗体浓度与 ADC 高度相关,可能无需进行该组分的 E-R 分析。
在进行剂量选择和策略考虑时,应综合考虑上述各方面的数据和分析。




ADC 药物的主要毒性和毒性来源
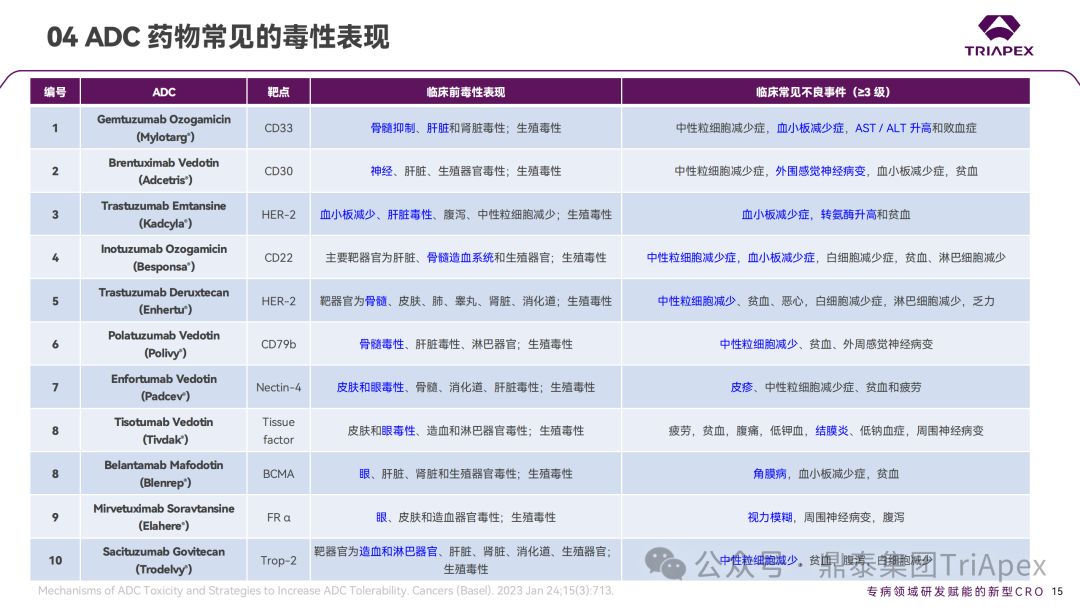
1. ADC 药物的主要毒性表现
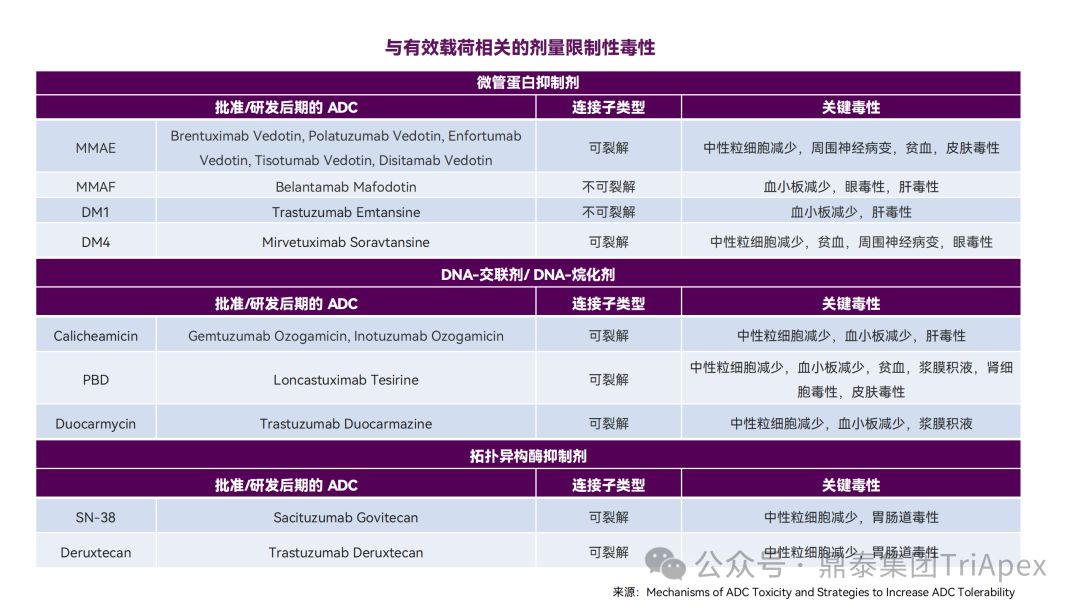
鼎泰团队根据 FDA 批准的 ADC 药物的说明书、审评报告和相关文献,对已上市的 ADC 药物在非临床研究和临床试验中常见的毒性表现进行了汇总。其中,血液学毒性是 ADC 药物最常见和最严重的不良反应之一。不同的 ADC 药物由于靶向抗原、payload 和偶联方式的差异,其毒性谱也有所不同。
2. ADC 药物主要的毒性来源
根据文献,ADC 注射给药后绝大多数会在非靶组织内脱偶联,导致非预期的毒性反应,最终只有约 0.1% 的 ADC 能够被递送至靶细胞中。
ADC 的毒性可能主要来自以下几种机制:
-
正常组织对 ADC 的非特异性摄取(靶抗原无关的脱靶效应)
-
抗体与正常组织中的抗原结合引起的肿瘤外在靶效应
-
具有细胞膜渗透性的 payload 产生的旁观者效应
-
连接子稳定性导致的细胞外脱偶联和 payload 释放导致的脱靶毒性
因此,ADC 的每个组分,包括抗体、连接子和 payload 都可能对 ADC 的安全性产生影响。
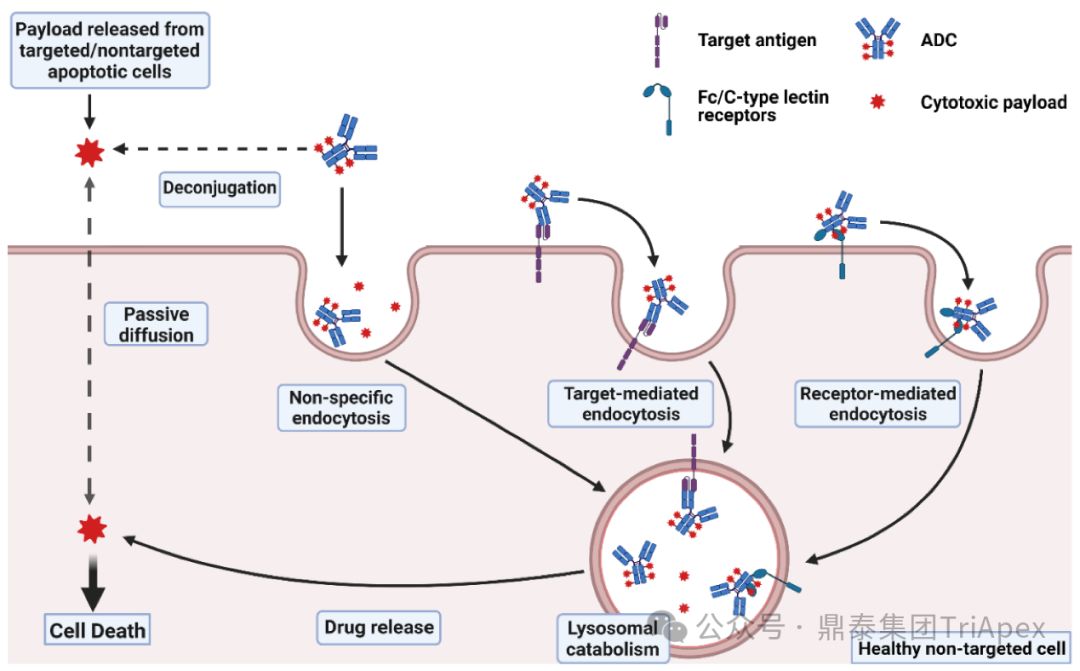
ADC 毒性机制[3]
ADC 药物临床剂量优化建议
1. 设计合理的 FIH 起始剂量
恰逢近期有业内资深同行对已上市 11 款 ADC 药物临床起始剂量设计进行推演,在征得原创作者同意的前提下,鼎泰团队根据原文内容进行了进一步的归纳整理(点击链接查看全文),在此感谢原文作者的辛勤努力和无私分享。
已上市的 ADC 药物均为肿瘤适应症。根据 2009年颁布的 ICH S9《抗肿瘤药物非临床评价》指导原则,推荐以啮齿类动物 STD10 的 1/10 和/或非啮齿类动物 HNSTD 的 1/6 作为起始剂量设计的依据。基于非临床研究数据,运用上述指导原则进行理论起始剂量估算,并与实际的 FIH 起始剂量进行对比,反向推演已上市 ADC 药物可能的起始剂量计算方法和可能的依据。
通过推演不难发现,大部分 ADC 药物采用 ICH S9 推荐方法计算的理论 FIH 起始剂量与实际采用的剂量相差不远。从计算过程来看,大部分产品进行了 BSA 转换(Padcev 除外)。推测原因可能是:ADC 药物活性高、毒性大,安全窗口通常较窄,采用 BSA 转换相对 BW 更为保守,也更为谨慎。这与单抗或其它安全性较好的生物制品 FIH 起始剂量的计算存在一定区别。
通过上述推演,不仅有助于我们更好地理解 ADC 药物非临床研究的试验设计的重点,也为未来新的 ADC 药物的临床起始剂量设计提供了有益的借鉴。
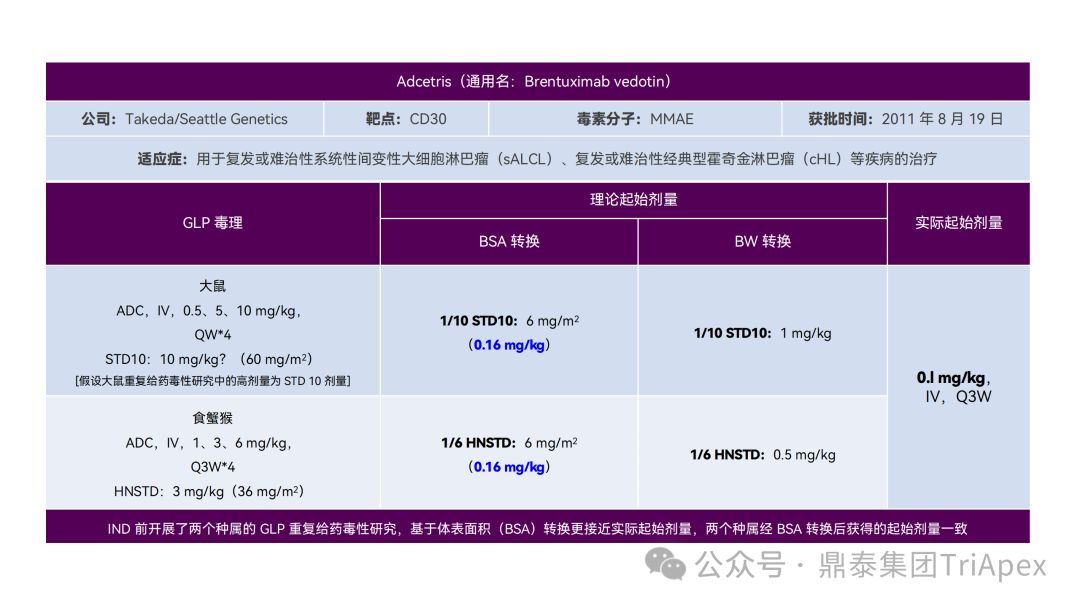

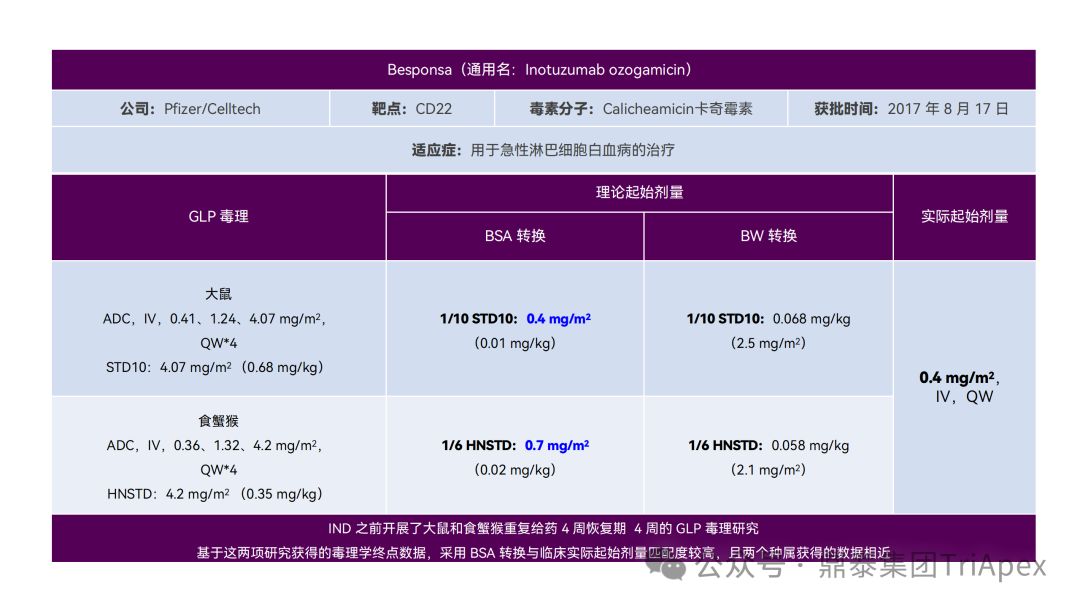

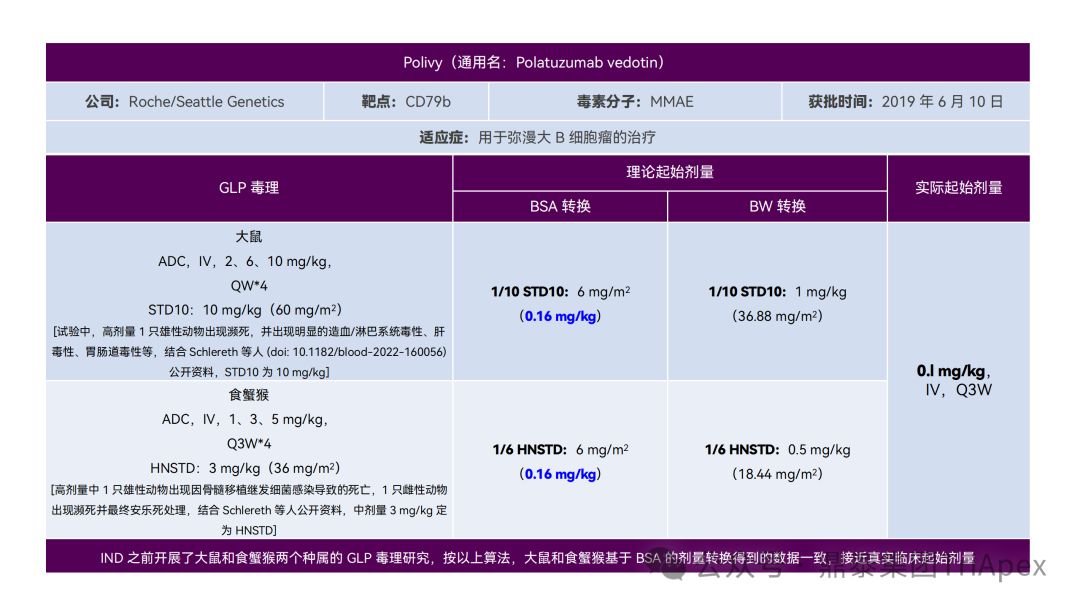
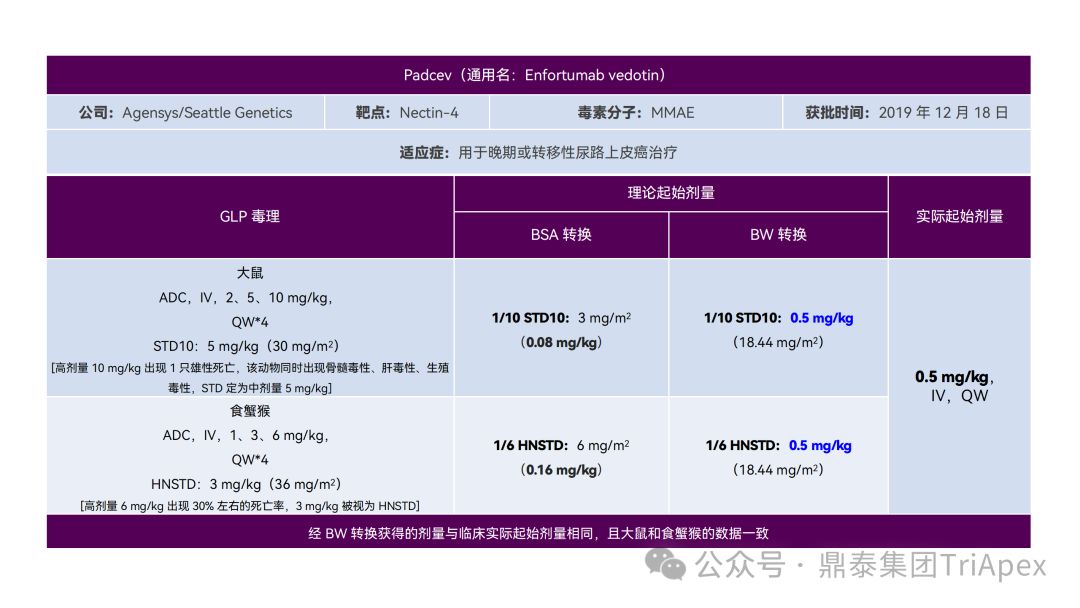
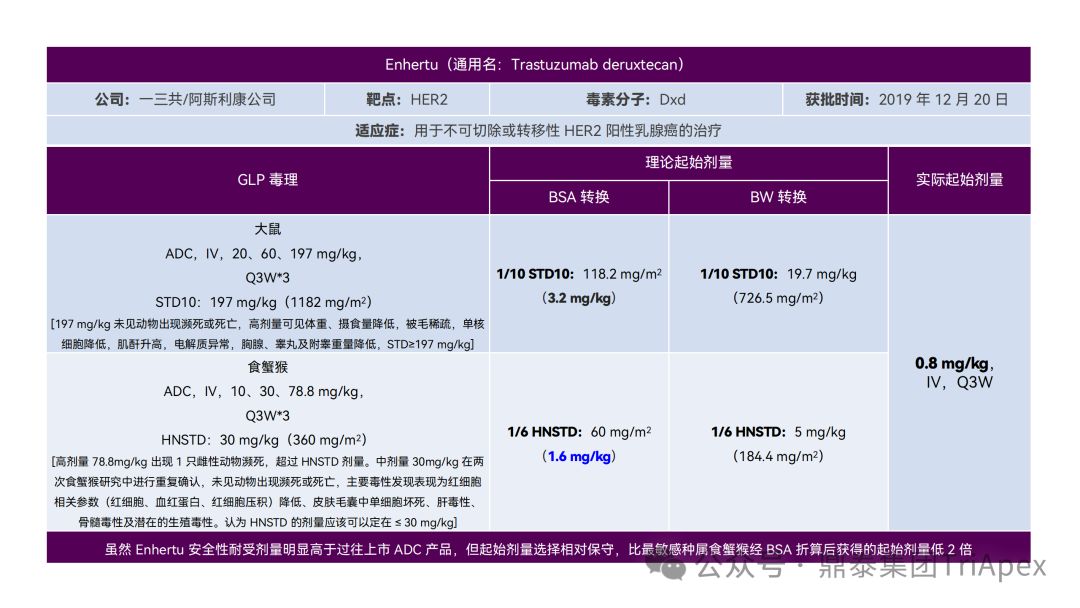
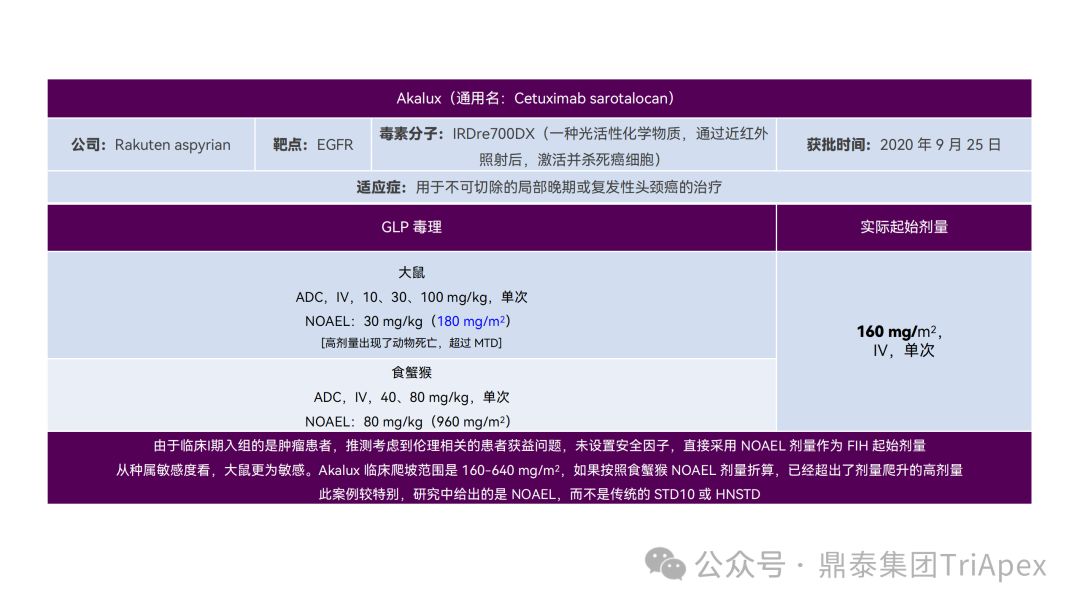


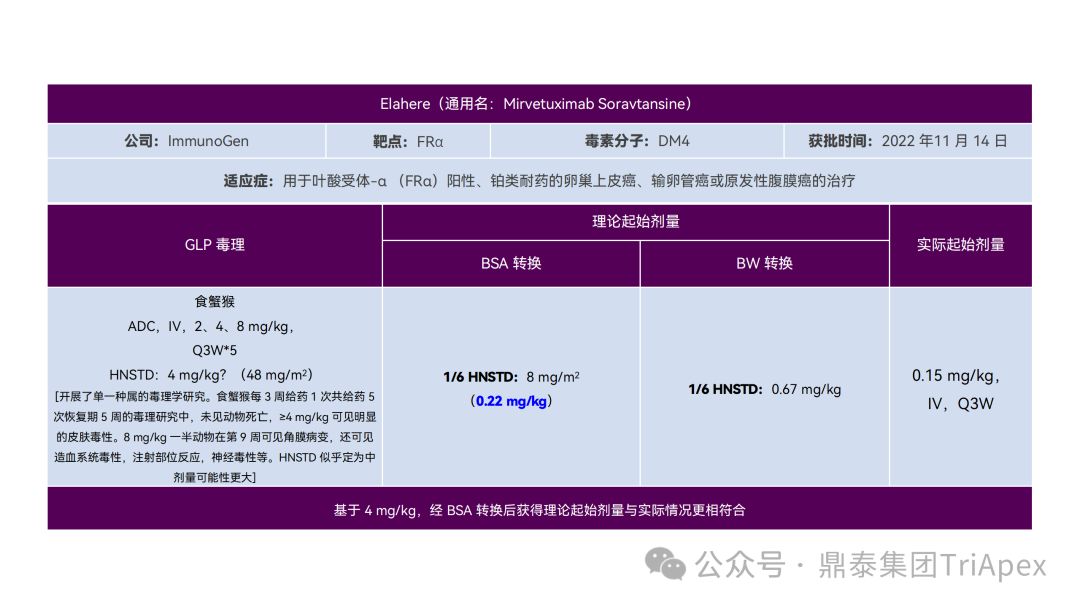
已上市11款 ADC 产品临床起始剂量设计推演
✦
左右滑动,查看更多
✦
2. 优化后续临床试验给药方案
-
设置基于体重的剂量上限(如 Adcetris),防止剂量过高和减少危重患者中的 AE
-
设定治疗期限(如 Polivy),有效缓解慢性 AE 的发生
-
优化给药频率(如 Mylotarg),以减少 Cmax驱动的 AE
-
根据患者相应情况调整剂量(如 Besponsa),适用于治疗窗较窄且响应时间较快的情况
-
以及随机剂量发现研究(如 Enhertu),可阐明不同剂量水平下的有效性和安全性
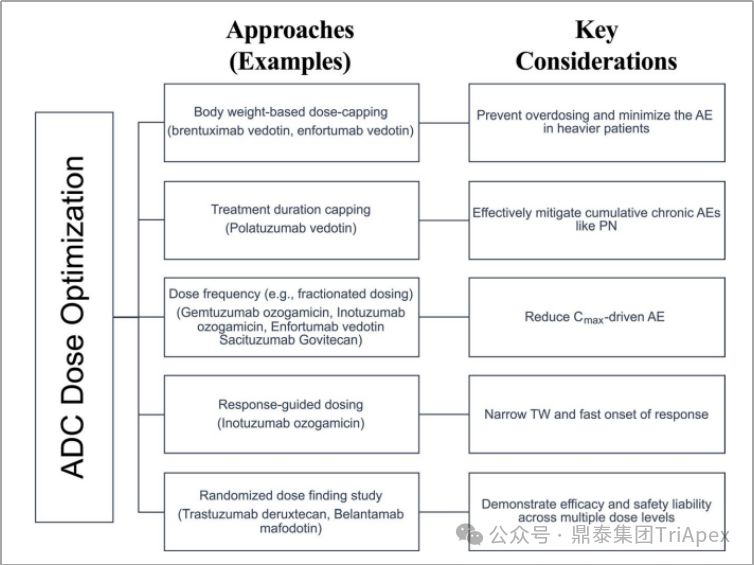
ADC 剂量优化策略和关键考虑因素[4]
2.1 设置剂量上限
根据患者体重调整 ADC 剂量可以实现剂量一致性,同时最大限度地减少个体间差异和毒性。
当体重对与时间无关的清除率和分布容积影响的幂指数在 0.5 左右时,建议考虑基于体重的剂量(mg/kg)。通常,当指数 <0.5时,固定剂量比基于体重的剂量带来的 PK 变异性更小;当指数>0.5 时,基于体重的给药比固定剂量带来的 PK 变异性更小。但基于体重的剂量也可能导致较重患者的暴露量高于平均水平。
患有糖尿病和肥胖等基础疾病的癌症患者可能更容易发生 AE。治疗期间常用的抗癌药和皮质类固醇可能会使糖尿病患者面临高血糖的风险,可能需要考虑调整剂量以减轻血糖升高的风险;对于肥胖患者,基于体重的给药容易导致剂量过度补偿。如 Adcetris(Brentuximab Vedotin, 维布妥昔单抗)和 Padcev(Enfortumab Vedotin,恩诺单抗)均以 BW 阈值 100 kg 进行剂量封顶,以降低个体间 PK 变异性和 AE 的潜在风险。
根据 Adcetris 的说明书,该药单药治疗时的用法用量为:1.8mg/kg,最大剂量 180mg,静脉输注 30 分钟, Q3W;对于既往未经治疗的 III 期或 IV 期经典霍奇金淋巴瘤(cHL)患者,联合化疗的推荐剂量为 1.2mg/kg,最大剂量为 120mg,Q2W,最多12剂。对于高危 cHL 和外周T细胞淋巴瘤(PTCL)患者,也设置了类似的剂量上限。一般来说,BW 上限可以提高 ADC 的安全性,并降低个体间暴露水平的变异性。体重大于 100 kg 的无体重剂量上限患者腹泻和疲劳发生率增加。


Adcetris 推荐剂量,资料来源:FDA Label 2023
2.2 设定治疗期限上限
设置治疗期限上限可以降低重复给药期间出现的慢性不良事件的风险,比如周围神经病变(PN)。研究发现,对于多种含 MMAE 的 ADC,如Polivy (Polatuzumab Vedotin,泊洛妥珠单抗)和 Adcetris(Brentuximab Vedotin,维布妥昔单抗), ≥2级 PN 发生率与偶联物(抗体偶联MMAE或偶联抗体)有关,与未偶联 MMAE 全身暴露无关[4]。
Polivy 由靶向 CD79b 的单抗 Polatuzumab、可裂解连接子 mc-vc-PAB 和 MMAE 偶联而成。通过 the type of parametric time-to-event (TTE)模型预测发现,Polivy 以 1.8 mg/kg Q3W,治疗 6 和 8 个周期时,≥2级PN 的发生率分别为 19% 和 31%,以 2.4mg/kg Q3W治疗时发生率分别为 27% 和 41%。此外,一项利用 8 种含 MMAE 的 ADC(约700 名患者)的独立分析发现,PN 风险随着偶联物暴露水平、治疗持续时间、体重和先前报道过 PN 而增加。由此可见,对给药剂量和治疗持续时间的调节可减少含有 MMAE 的 ADC 引起的 PN 风险。
上述分析支持 Polivy 在复发/难治性弥漫性大B细胞淋巴瘤(Untreated diffuse large B-cell lymphoma ,DLBCL)患者中以 1.8mg/kg Q3W持续给药6个周期的方案。类似的给药方案优化策略可能在未来进一步应用到其他 ADC 的延迟/慢性 AE 中。

Polivy 剂量和用法,资料来源:FDA Label 2023
2.3 优化给药频率
给药频率是另一个影响 ADC 安全性的重要因素。在相同的积累剂量(cumulative dose)下,优化给药频率可降低 Cmax,改善由 Cmax引起的不良反应。参考药代动力学(PK)和药效动力学(PD)信息来调整给药频率和治疗间隔,对于维持疗效和安全性至关重要,同时也提高了患者的便利性。
当将单个较高剂量分成多个较低剂量(如将9mg/kg Q3W分为3 mg/kg QW),每个周期产生相同的累积剂量、相似的累积(cumulative)AUC、较低的 Cmax 和更高的 Ctrough。因此,分次给药有望通过调节 Cmax 引起的毒性和/或 Ctrough 带来的疗效扩大治疗窗口期。
分次剂量可在给药周期内均匀分布。如果需要靶标介导的给药和/或较强的初始疗效,则可在较高的负荷剂量之后使用较低的剂量维持治疗;或者在治疗周期结束后加入给药间隔,以便毒性减弱。
在已批准的 ADC 中, Mylotarg(Gemtuzumab ozogamicin,吉妥珠单抗)最好地证实了优化给药频率的重要性。Mylotarg 由靶向 CD33 的单抗 Gemtuzumab、可裂解连接子 AcBut 和毒素 N-Acetyl Calicheamicin组成,是美国首个获批的 ADC。最初,Mylotarg 于 2000年被加速批准用于 60 岁以上且不适合化疗的复发性 CD33 阳性急性骨髓性白血病(AML)患者,给药剂量为 9mg/m2,IV,Q2W,共给药 2 次。在加速批准后的确证性III期临床试验(SWOG 106)中,Mylotarg 以 6mg/m2(使受试者 CD33 完全饱和的剂量)联合柔红霉素(NDR)/阿糖胞苷(AraC)治疗 60 岁以下的新发 AML 患者,疗效未优于标准治疗,且发现了严重的肝脏毒性和静脉闭锁性疾病风险,研究被提起终止。基于此,Mylotarg 于 2010年 撤市。
2017年,Mylotarg 重新获批上市。对于该过程,业内资深同行进行了深度解读(点击链接查看原文)。以下配图和注释引自原文:
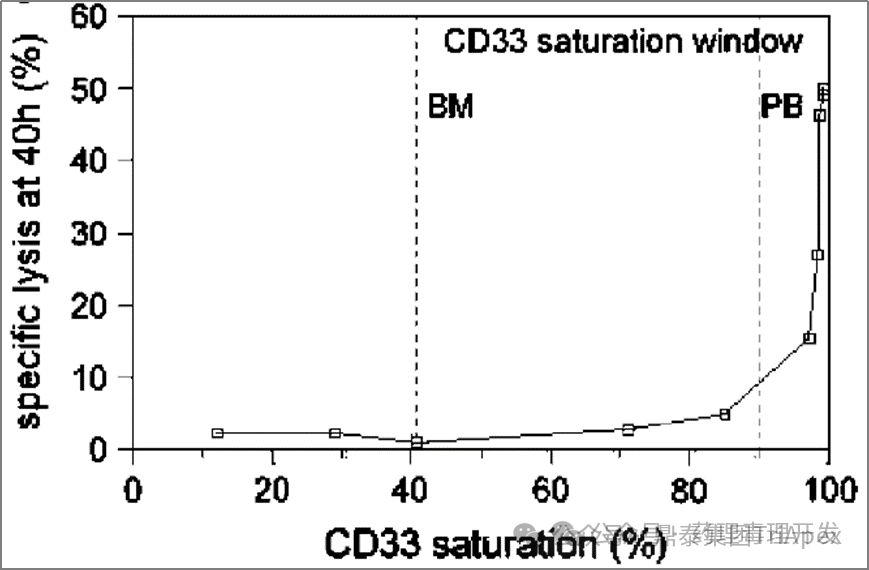
“而受体饱和与药效之间又有着很强的关联性。下图同样是采用患者 PB 和 BM 进行的研究,发现受体 >90% 饱和才能保障 GO 对细胞的有效杀伤。受体饱和程度<90%,GO 的杀伤活性仅剩 5% 不到。杀伤曲线非常陡峭。”
“以上研究结果说明,原来 9mg/m2 GO Day1 和 Day15 给药方案、或者 6mg/m2 Day4 给药方案,患者骨髓中的 CD33 受体是不饱和的,杀伤作用也可能是很有限的。因此,出现了一种新的研究思路,在饱和剂量下以更高频率给予 GO,比如首次药后,短时间内再次给药,可以快速饱和新生的CD33 抗原。另外,为了不增加毒性,考虑到 3mg/m2 即使 90% 以上受体饱和,可以进一步降低剂量。相当于把原来的 9mg/m2进行切割,总剂量不变,调整为短期内的多次 3mg/m2给药。基于这一优化的试验方案,先后启动了 3 项概念验证性临床研究(MyloFrance-1, Acute Leukemia French Association (ALFA)-0701, EORTC/GIMEMA AML-19)。”
MyloFrance-1 是一项开放、单臂 Ⅱ 期试验,入组 CD33 阳性首次复发的 AML 患者。诱导期接受 Mylotarg 单药 3mg/m2,IV,第1、4、7天给药。CR 患者使用阿糖胞苷巩固治疗。本试验中 CR 率与最初获得加速批准时的数据相当,且骨髓抑制的持续时间更短,无患者出现静脉闭塞性 AE。初步验证了低剂量、高频率给药方案的可行性。
在随后的 ALFA-0701 试验中,Mylotarg 以较低或分次剂量方案(3 mg/m2,IV,第1、4、7天给药)联合柔红霉素和阿糖胞苷,用于 50-70 岁的初发 AML 患者。疗效结果显示,该方案具有更长的中位无事件生存期 EFS (15.6月 vs 9.7月)和中位总生存期 OS(34月 vs 19.2月);安全性方面,试验组血小板减少发生率高于对照组,但未增加死亡风险。之所以有上述疗效和安全性的提升,主要原因在于通过以较低的剂量给药获得了比最初的给药方案(9mg/m2,Q2W)更低或更适度的 Cmax 和 AUC。受体占位试验显示,2mg/m2 或更高时,CD33 饱和度即可达到 90% 以上;而体内和体外研究表明,Mylotarg 给药后 CD33 迅速重新表达,这提示 Mylotarg 以更频繁的频率分次给药能获得更为有持久的 CD33 饱和且更高的安全性。
在另一项随机对照的III期临床试验(AML-19)中,入组了未经治疗且不适合化疗的 AML 患者。诱导期接受 Mylotarg 单药治疗,IV,第1天给予 6 mg/m2,第8天给予 3mg/m2;对照组给予最佳支持治疗。主要终点为 OS 显示了有统计学意义的显著获益(4.9月 vs 3.6月)。
基于以上临床试验结果,FDA 于2017年重新批准了 Mylotarg(3mg/m2,第1、4、7天)联合柔红霉素、阿糖胞苷治疗初治 AML;并批准了 Mylotarg 单药治疗初治且不适合化疗的 AML 成年患者(6mg/m2,第1天;3mg/m2,第8天),以及复发或难治性成人或儿童(2-17岁)AML 患者(3mg/m2,第1、4、7天)。与首次上市时采用的 9mg/m2 Q2W方案相比,优化后的给药方案提高了 Mylotarg 的获益-风险比。
Mylotarg 剂量优化和获得监管机构重新批准上市还涉及很多值得深入探究的多学科综合知识的运用。曾子像老师的一篇推文给出了非常精细和到位的分析,详细点击链接查看(PK/PD 助力 ADC 产品 Gemtuzumab ozogamicin 起死回生—临床设计一念天堂一念地狱)。在此特别感谢曾子老师的总结和指引。
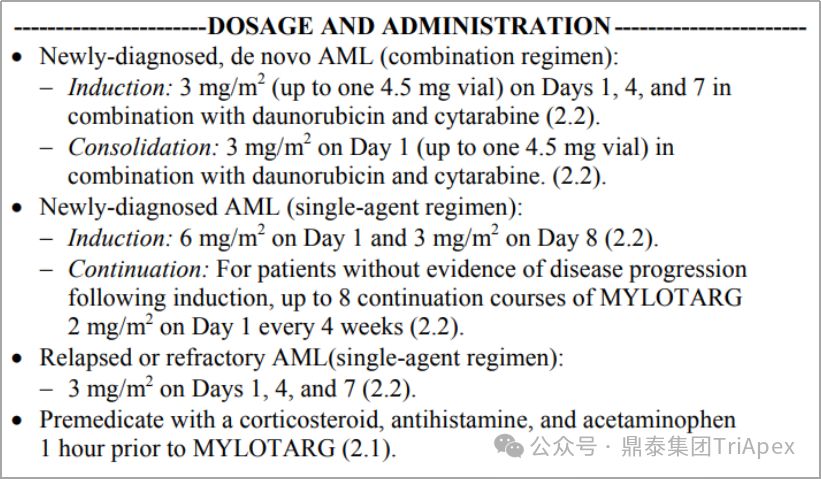
Mylotarg 剂量和用法,来源:FDA label 2017
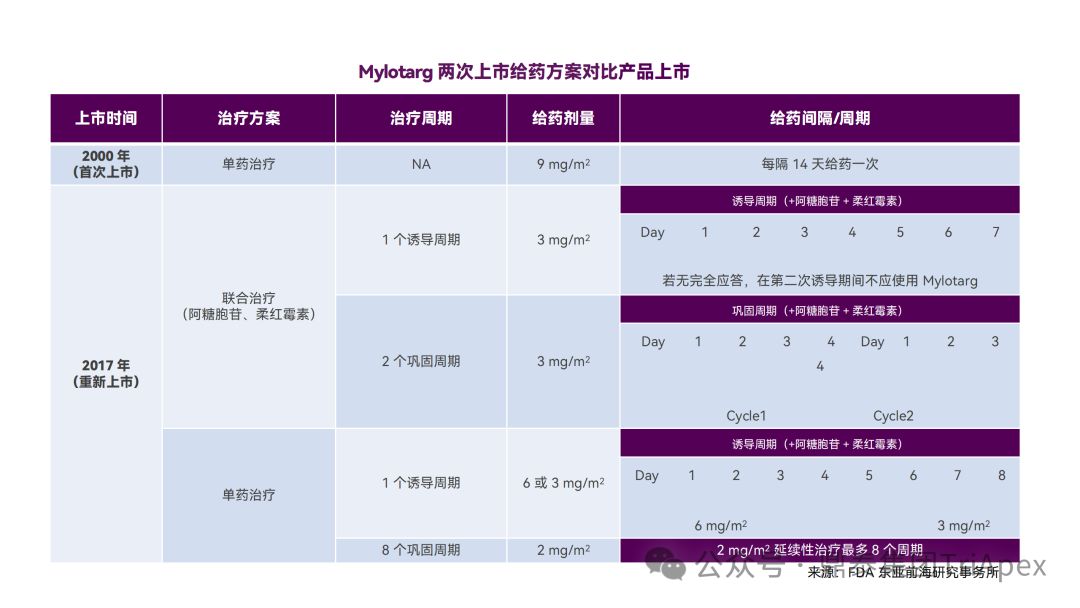
2.4 根据治疗响应情况调整剂量
根据患者的治疗响应情况调整剂量是一种适应性给药策略。Besponsa (Inotuzumab ozogamicin,奥加伊妥珠单抗)由靶向 CD22 的单抗 inotuzumab、酸可裂解连接子 AcBut 和毒素 Calicheamicin 组成。Besponsa 采用基于早期疗效的适应性给药方案,以应对其毒性(如血小板减少、静脉阻塞)和治疗响应驱动的非线性PK(如 CR/CRi 患者暴露量增加)。第1周期(21天为一个周期)的初始剂量为 1.8mg/m2,分 3 次给药;后续周期的给药方案根据响应情况调整为:
-
如患者达到 CR 或不完全恢复(Incomplete Count Recovery,CRi),则调整为每周期(28天为一个周期)1.5mg/m2,仍然分3次给药
-
如未达到 CR/CRi,则恢复至每周期(28天为一个周期)1.8mg/m2
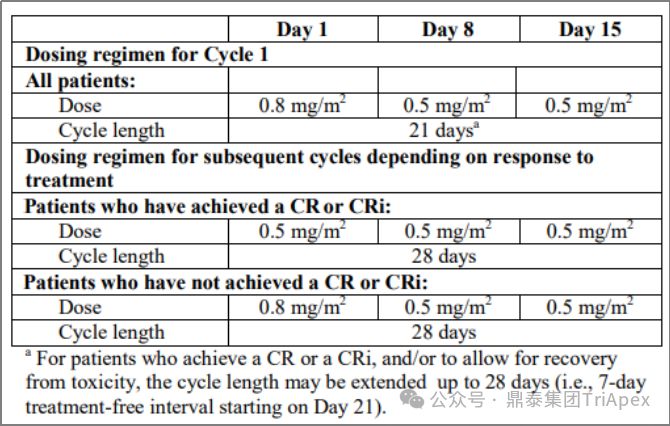
Besponsa 剂量和用法,来源:FDA label 2017
降低剂量的基本原理是有助于降低毒性,因为在 CR/CRi 患者中观察到较高的 Besponsa 暴露,这表明肿瘤负荷或细胞计数可能影响 Besponsa 消除(即响应依赖性 PK)。在临床试验中,在治疗周期的后期进行剂量调整是非常常见的,大多数患者在每周期给予 1.8mg/m2后出现 AE,导致给药延迟(78%)或降低剂量(22%)。
2.5 随机剂量探索研究
II/III期临床试验通常不提供多种剂量水平的有效性和安全性数据,这是肿瘤学和免疫肿瘤学剂量优化的主要障碍。理想情况下,使用多个随机剂量水平和足够大的样本量的前瞻性研究可最大限度地了解多个有效剂量水平的获益-风险特征及其 E-R 关系,这将为剂量选择和优化提供关键支持。
FDA 批准的两个 ADC,Enhertu(Trastuzumab deruxtecan,德曲妥珠单抗)和 Blenrep(Belantamab mafodotin,玛贝妥单抗)在II期临床试验中采用了该策略。
为确定 Enhertu 在 HER2 阳性、不可切除或转移性乳腺癌中的推荐剂量,在一项II期临床试验中,患者按 1:1 随机接受 5.4mg/kg 或 6.4mg/kg治疗。2个剂量组的 ORR 分别为 52.6%(20/38)和 55.7%(34/61),模型预测的6个月 PFS 率分别为 87% 和 90%。总体而言,6.4mg/kg 剂量预计具有更好的疗效,但也会增加发生 TEAE 或因 TEAE 而停止用药/减少剂量的风险。根据 E-R 关系和 PK 分析预测的获益-风险概况,选择 5.4mg/kg Q3W作为推荐剂量。

鼎泰集团多年来专注于 ADC 药物的非临床评价和临床研究,已积累了近 50 多个 ADC 药物的研究经验,并为我们的合作伙伴快速推进其 ADC 药物确定 PCC 和进入临床试验奠定了坚实的基础。
这些产品中所涉及的靶点涵盖 HER2、Trop-2、DLL3、CLDN18.2、EGFR、FRα、B7-H3、EGFR/TAA 等热门靶点,以及 SN-38、Dxd、Exatecan、MMAE 和艾日布林等主流的毒素分子。
鼎泰集团始终坚持以科学性为前提、药政策略为导向,为 ADC 药物从分子发现、非临床研究(支持 IND 和 NDA)、临床开发(早期临床、剂量优化、关键性临床)以及注册申报提供一体化深度赋能。
鼎泰智高点 - ADC 热文 & 典型项目案例
● 靶向 FRα 的 Mirvetuximab soravtansine (Elahere) 的上市路径回顾和对 ADC 药物非临床开发的启示
● 全球已上市 ADC 药物非临床研究路径调研-拓扑异构酶抑制剂篇
剂量优化图破壁,分次给药有创意。
转化研究好魄力,建模模拟有依据。
曾子老师有主意,相得益彰真情谊。
获益风险两相依,临床价值立天地。
参考资料:
[1] FDA.Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases Guidance for Industry
[2] FDA.Clinical Pharmacology Considerations for Antibody-Drug Conjugates Guidance for Industry
[3] Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. https://doi.org/10.3390/cancers15030713
[4] Liao MZ, Lu D, Kågedal M, Miles D, Samineni D, Liu SN, Li C. Model-Informed Therapeutic Dose Optimization Strategies for Antibody-Drug Conjugates in Oncology: What Can We Learn From US Food and Drug Administration-Approved Antibody-Drug Conjugates? Clin Pharmacol Ther. 2021 Nov;110(5):1216-1230. doi: 10.1002/cpt.2278. Epub 2021 Jul 8. PMID: 33899934; PMCID: PMC8596428.





免责声明:本文来自鼎泰集团内容团队,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台,如需转载请添加微信LXL--7。本文仅作信息交流而非商业盈利之目的,内容仅供分享学习。